

Theranos, the much-hyped Silicon Valley startup valued at an eye-popping $9 billion, has been facing questions over whether its proprietary technology can actually do what it claims: diagnose diseases and health conditions from a few drops of blood.
The Wall Street Journal first raised those questions in an Oct. 16 story, followed by a report that Theranos recently stopped collecting tiny vials of blood for all but one of its tests after a recent FDA inspection. Theranos has defended itself, saying that the stories "say or imply many things that are simply wrong"; the WSJ has stood by its reporting.
The saga took another twist Tuesday when the FDA posted documents from that inspection during late August and September in response to public records requests. As heavily redacted and jargon-filled as they are, they don't look great for Theranos: The FDA questioned whether its technology was validated, called its blood vial "uncleared," and said the company wasn't set up to handle customer complaints or vet its internal processes.
1. The FDA questions whether Theranos' technology works.
The agency argued that Theranos failed to show adequate proof, in real or simulated conditions, that its technology lived up to what it claimed to do.
"Given the number of observations that relate to the same sorts of topics, I think it's fair to say generally that the FDA is concerned about the validation under the appropriate conditions of the design" of the technology, Patti Zettler, an associate professor of law and FDA expert at Georgia State University, told BuzzFeed News.
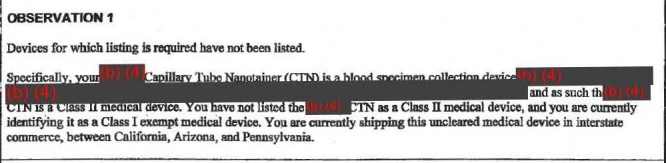
The agency also said that Theranos' "nanotainer" — its tiny vial for drops of blood — isn't cleared by the FDA or properly classified according to FDA definitions, yet Theranos was sending them throughout California, Arizona, and Pennsylvania. "If that observation holds, that's a pretty clear violation of the law," Zettler said.
The documents:


2. The FDA says Theranos wasn't set up to handle complaints about its products.
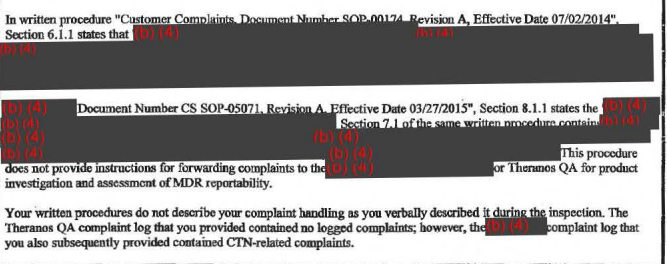
The company doesn't have a satisfactory procedure for receiving, reviewing, and assessing complaints, and its written description of that process didn't match what Theranos verbally told regulators during their visit, according to the FDA.
In at least one case, the agency says, this led to Theranos failing to investigative, or even acknowledge, a complaint. The complaint concerned "difficulties in inspecting [nanotainer] specimen quality" (presumably, the blood inside the nanotainer). Some aspects of it were apparently "too opaque to be able to see clotting clearly."
The documents:


3. The FDA questions Theranos' internal quality control system.
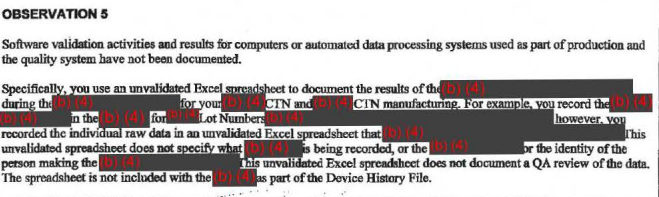
The FDA wasn't happy with the way Theranos was vetting components of its business — like its suppliers' qualifications and the software and data-processing systems used in production — saying they weren't properly documented or audited.
The documents:



These documents are what's known as Form 483s, which FDA inspectors issue if they see anything that may violate the federal Food, Drug, and Cosmetic Act and related laws. They're not final determinations of whether they're violations, though, and the company is given a chance to correct them.
"This is not the equivalent of a warning letter," Zettler told BuzzFeed News. "Keeping in mind that these are preliminary documents, I think the overall feeling is that something's going on, it's just not entirely clear what, and what's going to happen from here is an open question."
In response to our voluntary outreach to the FDA in the fall of 2013, FDA's guidance was that they wanted to regulate our Nanotainer tubes and review our proprietary systems, and we submitted them for clearance. We then worked closely with FDA, culminating in our first FDA clearance for HSV-1 in July, which included clearance of the associated proprietary Theranos devices, software and Nanotainer tubes for that test. Our first FDA inspection began on August 25, 2015, as we have previously explained. This inspection focused on Theranos' FDA quality systems compliance, on which we had been working previously, so as to transition from the CLIA laboratory quality systems to the FDA framework. We worked closely with FDA representatives and provided all requested information. At the conclusion of the inspection, FDA issued two Form 483s outlining observations made during the inspection. We believe that we addressed and corrected all the observations at the time of, or within a week of, the inspection and have submitted documents to FDA that say so, including extensive documentation, as well as outlining our path to the whole QSR transition going forward. Their observations are, by their very nature, preliminary and in this is a time of evolving policy, we have advocated for the transition from the LDT framework to the FDA quality systems framework. Thus, we voluntarily decided to transition in real-time to QSR in all areas, including prior to certain clearances despite the evolving policy in this space. We are charting new pathways, and are working to create a model for the transition. In our discussions with FDA, we determined that in order to do so, it was appropriate to temporarily pause use of the Nanotainer™ tubes for all tests except our cleared HSV-1 test as we cut over to FDA quality systems, and wait for clearance. This transition is part of converting all our operations from the LDT framework to the FDA framework. We believe this is the right action to take as we wait for clearance of the Nanotainer™ tubes, for which we have submitted all our data to the agency, so that we incorporate them into other tests under the FDA quality systems framework upon clearance. We are in close contact with FDA in this process. Over 120 of the tests developed for use on our devices used as part of Theranos proprietary technologies have been submitted in pre-submissions to FDA. This voluntary engagement with FDA is not only a statement about our confidence in our technologies, but this summer's clearance and CLIA waiver would not be possible if our devices and technologies did not work with those tests. Theranos' technology is much more than the Nanotainer tube. Our proprietary systems and technologies – which we protect as intellectual property – have been rigorously tested and reviewed.

UPDATE
This story has been updated to include comments from law professor Patti Zettler.